
Recently, the team led by Assistant Professor Ren Yuxiang from Nanjing University, in collaboration with Professor Huang Wenbing and Professor Wen Jirong from Gaoling School of Artificial Intelligence, Renmin University of China, as well as researchers from Huawei Central Research Institute, proposed DAO (Diffusion-based Crystal Omni), the first siamese foundation model framework for crystal structure prediction. This work has been accepted for publication in Nature Communications, a top-tier international academic journal.
This framework aims to address a fundamental and highly challenging task in the field of materials discovery—predicting stable three-dimensional crystal structures solely from chemical compositions. Due to the extremely complex three-dimensional geometric configurations of crystals, traditional prediction methods based on first-principles calculations (DFT) or evolutionary optimization often suffer from serious bottlenecks, such as high computational costs and poor scalability with system complexity. Although deep generative models have been introduced to this field, existing models primarily rely on small-scale, domain-specific datasets for training, resulting in limited generalization ability to unknown structures. The research team has broken through the limitations of current approaches and innovatively constructed a new training paradigm.
The overall framework consists of two complementary siamese foundation models working in synergy: a generator (DAO-G) responsible for generating stable structures, and a predictor (DAO-P) focused on energy prediction and auxiliary generation. The models are first pre-trained on a massive dataset (CrysDB) containing approximately 940,000 stable and unstable crystal structures with energy annotations, and can then be fine-tuned for precise application to specific downstream tasks.
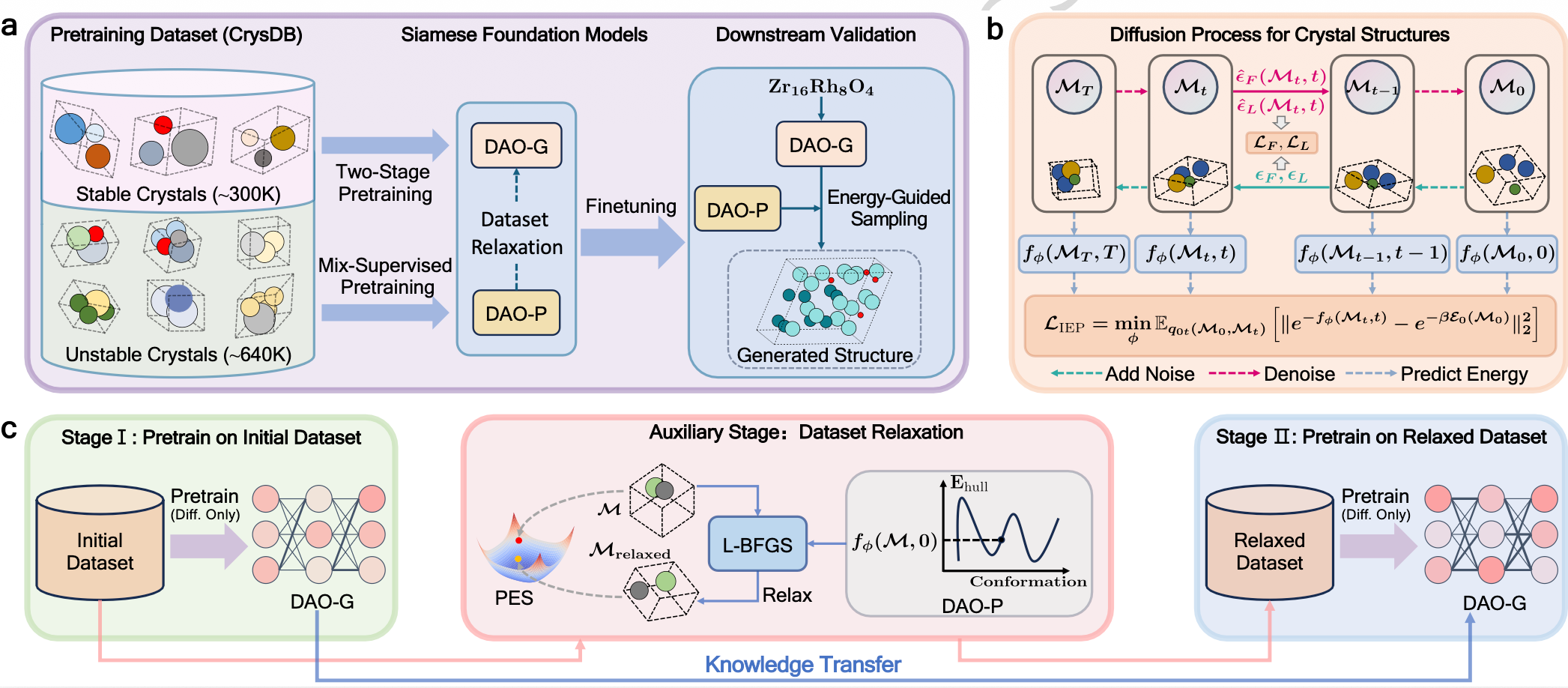
Figure 1: Schematic diagram of the pre-training, fine-tuning, and inference pipeline of the DAO framework
In terms of specific mechanism design, the DAO framework serves as a bridge connecting massive data with stable structure generation, incorporating three core innovations:
Geometric Architecture Optimization. Both DAO-G and DAO-P are built upon the novel geometric graph, Crysformer, proposed by the team. This architecture effectively characterizes the geometric features of input crystals and rigorously guarantees the equivariance and periodic translation invariance required for predicting crystal lattices and coordinates.
Two-Stage Pre-training with Dataset Relaxation. In the first stage, DAO-G is pre-trained on the full CrysDB dataset containing a large number of unstable structures. In the second stage, DAO-P serves as an efficient energy predictor to replace expensive DFT calculations, computing energy gradients to relax high-energy unstable structures and optimize them into more stable configurations, thereby eliminating the generation bias introduced by unstable data.
Hybrid Supervised Pre-training with Energy-Guided Sampling. DAO-P is pre-trained by combining self-supervised diffusion loss with exponential energy loss, enabling accurate estimation of intermediate state energies along the diffusion generation trajectory. During the sampling phase of structure generation by DAO-G, DAO-P is introduced as an energy guide with energy gradient guidance, improving the thermodynamic stability of the final generated crystals and the matching rate of complex structures.
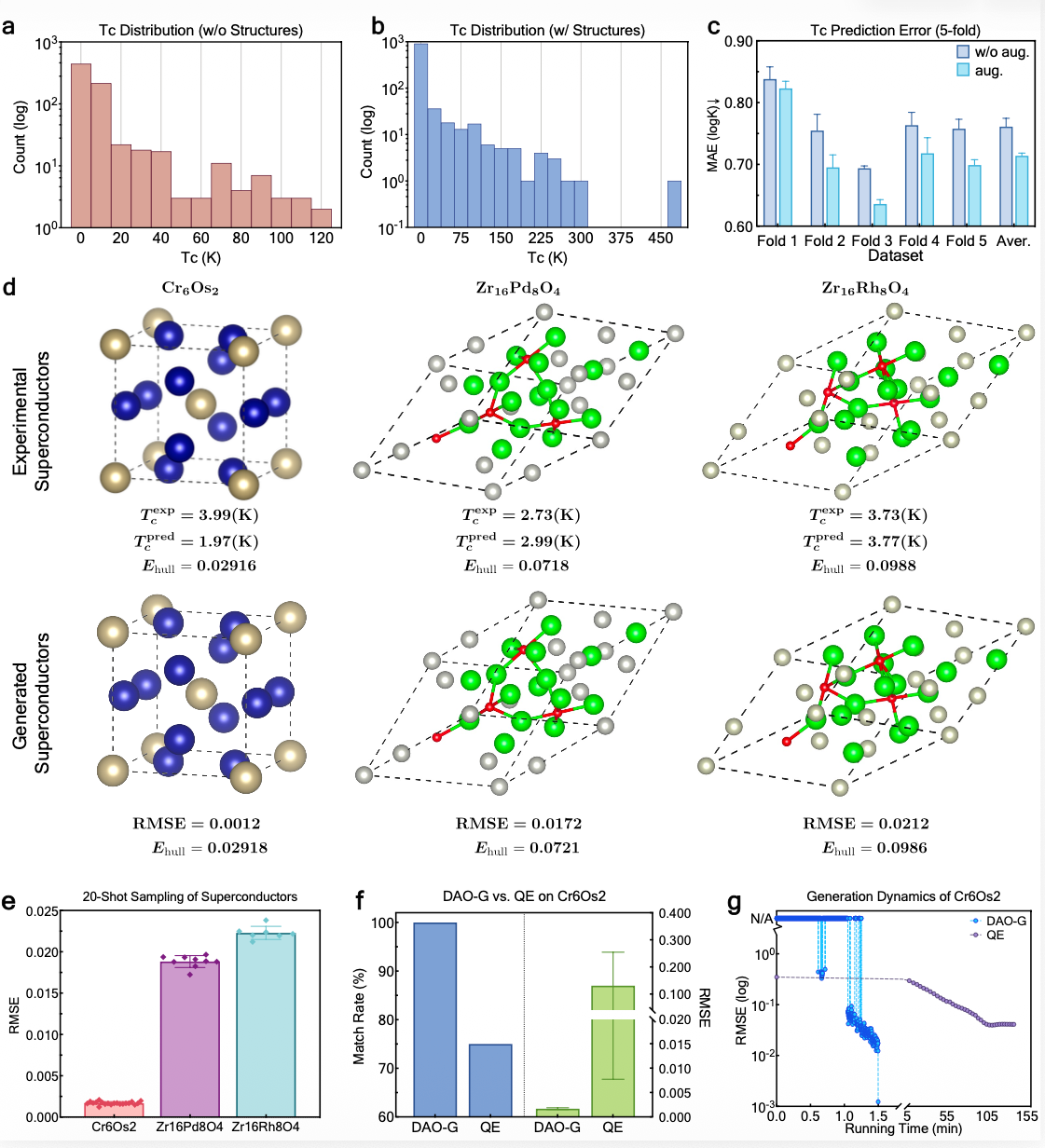
Figure 2: Visualization of structure prediction results of DAO on three real superconducting materials and efficiency comparison with traditional DFT methods
The results demonstrate that the DAO framework significantly improves prediction performance on two established CSP benchmarks, exhibiting strong effects across various skeleton architectures. More remarkably, when tested on three real-world superconducting materials (Cr₆Os₂, Zr₁₆Rh₈O₄, and Zr₁₆Pd₈O₄) that are notoriously difficult for traditional computational methods, DAO showed overwhelming advantages. Taking Cr₆Os₂ as an example, DAO achieved a 100% matching rate with the experimental reference structure across 20 generation trials, with an atomic position root-mean-square error (RMSE) as low as 0.0012, while delivering a computational speedup of over 2,000 times per iteration compared to traditional DFT-based structure predictors. These compelling results not only provide a novel and efficient tool for the design of complex polymorphic and superconducting materials but also strongly underscore the immense potential of AI foundation models in advancing cutting-edge materials science research.